|
Темы сайта |
|
  |
|
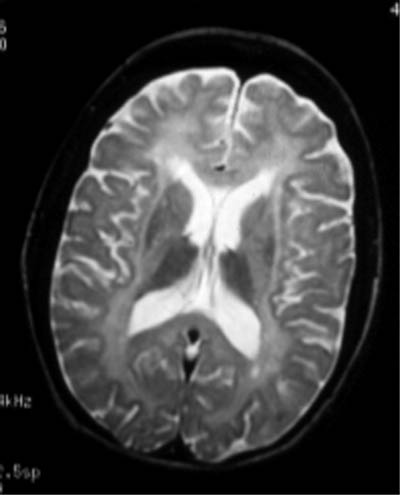
Лейкодистрофия - группа наследственных заболеваний
нервной системы, при которых нарушается процесс миелинизации
вследствие генетически обусловленого энзимного дефекта. Для всей
группы лейкодистрофии характерно начало в детском, реже в
юношеском возрасте, прогрессирующее течение с присутствием в
клинической картине в качестве ведущих симптомов психической
деградации, снижения зрения и спастических парезов; в
терминальной стадии развивается традиционно синдром
децеребрационной ригидности. При морфологическом исследовании
обнаруживаются симметричные, диффузные, слабо отграниченные
области распада миелина в полушариях мозга и мозжечка. Продукты
распада липидов миелина накапливаются в тканях мозга и
внутренних органах. Аксоны в очагах гибели миелина и ганглиозные
клетки содержат продукты нарушенного метаболизма миелина.
Основными формами лейкодистрофий являются: метахроматическая
лейкодистрофия Шольца-Гринфилда, глобоидно-клеточная
лейкодистрофия Краббе, лейкодистрофия Галлевордена-Шпатца,
суданофильная лейкодистрофия Пелицеуса-Мерцбахера, спонгиозная
дегенерация белого вещества мозга (болезнь Канавана-ван
Богарта-Бертрана), лейкодистрофия с наличием диффузной
волокнистой формации Розеиталя (болезнь Александера). |
Метахроматическая лейкодистрофия Шольца-Гринфилда
|
Заболевание характеризуется диффузными очагами демиелинизации. В
головном мозге, в белом веществе мозга и периферических нервах
обнаруживаются метахроматические вещества, представляющие
сульфатидные липиды. Аналогичные вещества могут выявляться в
нервных клетках мозга, сетчатке глаза, внутренних органах. При
метахроматической лейкодистрофий установлена инактивация энзима
арилсульфатазы А, что приводит к серьезным нарушениям в обмене
сульфатидов. Арилсульфатаза А представляет термолабильную
фракцию цереброзидной сульфатазы. Клинически заболевание
характеризуется возникновением первых симптомов в возрасте 2-3
лет. Обнаруживаются спастический парапарез или тетрапарез,
атактический синдром, судороги. Прогрессирует снижение
интеллекта, нарушается речь; зрение и слух снижаются. Позднее
выявляются бульбарные и псевдобульбарные симптомы, тетраплегия,
децеребрациональная ригидность. В ликворе обычно
белково-клеточная диссоциация. При дополнительных исследованиях
обнаруживаются диффузные изменения на ЭЭГ, метахроматические
включения при биопсии периферических нервов. Характерным
признаком является снижение или отсутствие сульфатазы А в моче,
положительный тест при окрашивании осадка мочи толуидиновым
синим. Указанные признаки подтверждают диагноз метахроматической
формы лейкодистрофии.
Дифференциальный диагноз проводится с детским церебральным
параличом. Неуклонное течение заболевания с присоединением новых
симптомов и дополнительные специфические признаки позволяют рано
ставить правильный диагноз. Прогноз при метахроматической
лейкодистрофии неблагоприятный: больные погибают спустя
несколько лет, чаще в возрасте 3-7 лет, от интеркуррентных
инфекций. |
Глобоидно-клеточная лейкодистрофия Краббе
|
Основные изменения обнаруживаются в обмене цереброзидов,
следствием чего является диффузный склероз мозга. В отличие от
болезни Гоше цереброзиды при глобоидно-клеточной лейкодистрофии
в качестве основного углеводного компонента содержат галактозу.
Страдает преимущественно белое вещество головного и спинного
мозга. В свежих участках демпелинизации отмечается скопление
глобоидных клеток, клетки крупные, много-ядерные, содержат
большое количество цереброзидов. Клиническая картина заболевания
характеризуется началом в грудном возрасте (4-5 месяцев и
позже). Проявляется раздражительностью, плаксивостью ребенка,
приступами крика и судорог. В неврологическом статусе отмечается
дистония мышц с тенденцией к гипертонии, позднее мышечная
ригидность, бульбарный синдром, нарастает деменция, снижение
слуха. На глазном дне обнаруживается атрофия сосков зрительных
нервов. В ликворе умеренно повышается количество белка.
Диагноз глобоидно-клеточной лейкодистрофии основывается на
развитии заболевания в раннем детском возрасте, сочетании
указанных выше клинических симптомов и неуклонном
прогрессировании процесса. Течение болезни крайне
злокачественное, быстро наступает летальный исход. |
Лейкодистрофия Галлевордена-Шпатца
|
|
Патологоанатомически заболевание характеризуется
преимущественным поражением стриопаллидарной системы. Клетки
бледного шара и черного вещества содержали большое количество
железосодержащего пигмента. Менее выраженные изменения
наблюдались в ганглиозных клетках других отделов мозга,
выявлялись очаги демиелинизации. Данные явления свидетельствуют
о значительных нарушениях пигментного и липидного обмена. Первые
симптомы заболевания обнаруживаются в возрасте 7-12 лет:
возникают полиморфные гиперкинезы мышц лица, туловища и
конечностей. В последующем отмечаются ригидность мышц,
замедление темпа и ограничение объема движений, атактический
синдром, деменция, иногда судороги, в поздних стадиях -
бульбарные расстройства. Заболевание медленно прогрессирует,
длительность его составляет от 10 до 30 лет. |
Суданофильная лейкодистрофия Пелицеуса-Мерцбахера
|
|
Морфологически выявляется диффузная демиелинизация головного и
спинного мозга при относительной сохранности осевых цилиндров,
что обусловливает пестроту гистологической картины.
Лейкодистрофия отличается началом в возрасте 5-10 месяцев и
медленным прогрессированием. Чаще поражаются мальчики.
Характерными симптомами являются нистагм (горизонтальный,
вертикальный и ротаторный), дрожание головы, координаторные
расстройства. По мере течения заболевания повышается мышечный
тонус, снижается интеллект, обнаруживаются гиперкинезы или
паркинсоноподобный синдром, атрофия зрительных нервов. В ликворе
может несколько повышаться количество белка и клеток. Болезнь
довольно быстро прогрессирует в первые годы, но в дальнейшем
могут наблюдаться ремиссии, течение становится медленным, иногда
наблюдаются стационарные формы. Больные иногда доживают до 30-40
лет. |
Спонгиозная дегенерация белого вещества мозга (болезнь
Канавана-ван Богарта-Бертрана)
|
|
Болезнь отнесена к лейкодистрофиям вследствие наследственной
природы заболевания и преимущественного поражения белого
вещества мозга. Мозговая ткань резко гидрофильна, наблюдается
значительная демиелинизация. В демиелинизированной ткани резко
снижены или отсутствуют фосфолипиды, цереброзиды и сфингомиелины.
Возможно нарушение процессов миелинизации еще при внутриутробном
развитии ребенка. Мальчики болеют чаще девочек. В большинстве
случаев при рождении ребенка отмечаются адинамия и анорексия,
часто клонико-тонические судороги. Спустя несколько месяцев
выявляется снижение тонуса мышц шеи и повышение его в
конечностях, что придает своеобразную позу больному. Из других
симптомов следует назвать гидроцефалию, атрофию сосков
зрительных нервов, гиперкинезы, глазодвигательные расстройства,
деменцию. Довольно быстро теряется слух, зрение. Возникает
состояние децеребрационной ригидности, картина бульбарного
синдрома. При люмбальной пункции могут быть отмечены повышение
давления ликвора, увеличение белка. На краниограмме - явления
гидроцефалии. Летальный исход наступает до 2 лет. |
Лейкодистрофия с наличием диффузной волокнистой формации
Розеиталя (болезнь Александера)
|
|
Крайне редкое заболевание. Характеризуется диффузными
скоплениями гиалина в мозговой ткани, представляющими продукты
распада миелина. Весьма возможно, что изменения обмена миелина
вторичны и происходят вследствие нарушений формирования
астроцитами глиальных фибрилл. Клиническая картина: заболевание
возникает в грудном возрасте, отмечаются гидроцефалия,
судорожные приступы, слабоумие, двигательные расстройства. |
Лечение лейкодистрофий
|
|
Лечение лейкодистрофий до настоящего времени не эффективно.
Применяется симптоматическая терапия: дегидратационные и
противосудорожные средства, назначаются препараты, снижающие
мышечный тонус, и др. Делаются попытки применения препаратов,
влияющих на обмен веществ: ферментов, гормонов и т. д. При
проведении медико-генетической консультации оправданы
рекомендации о прекращении дальнейшего деторождения в семьях,
где имелись случаи лейкодистрофии. Прогноз неблагоприятный,
летальный исход нередко наступает в первые годы жизни. |
|
По материалам:
http://cgenetic.ru/. |
|
